JHZR2
Staff member
This is an analysis of my own make (mods, move it somewhere else if you desire), which hopefully will find some interest on BITOG. Im doing gas chromatography analysis of various fuels for projects that Im working on, and so am quantifying various fuels and additives via flame ionization detection and pulsed flame photometric detection. The FID is a hydrocarbon specific destructive detector - it separates the hydrocarbons through the power of chromatography, then burns them to classify. FID results will be my first set of posts.
Some might be interested by this, I hope.
First, gasoline:
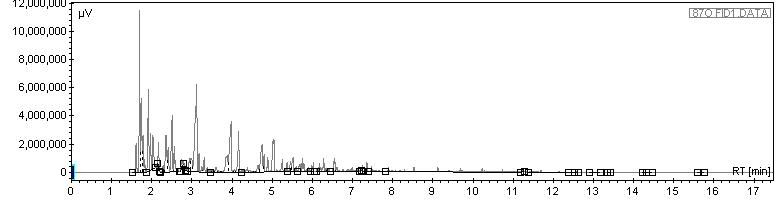
Next, gasoline doped with a few drops of n-octane. Note that due to all the light boilers, there are a lot of peaks on the far left that dont separate; it is very difficult to resolve all the hydrocarbons in gasoline, plus, given the overwhelming presence of C8 species, its tough to see a difference:

Next, a little heavier - JP-5 fuel, which is a high flashpoint Navy jet fuel:

Next, JP-5 fuel doped with some n-c8. Since chromatography is a comparative technique at heart, the addition of the notable c8 spike lets you recognize the rest of the hydrocarbons:

Next, road diesel fuel. Since all of these fuels were tested by the same chromatography method, it is valid to compare the times it takes on the scale for the various compounds to elude on these chromatograms versus those of the other fuels:

Next, road diesel fuel doped with some n-c8 for numbering purposes. Fortunately this method doesnt separate the isomers of the various hydrocarbons, so we get real nice distinct peaks where each one is:

And, once again, since it is a comparative technique and the same method was always used... here is the general location where your C8 molecules come through:

Enjoy!
[ July 18, 2006, 04:59 PM: Message edited by: JHZR2 ]
Some might be interested by this, I hope.
First, gasoline:
Next, gasoline doped with a few drops of n-octane. Note that due to all the light boilers, there are a lot of peaks on the far left that dont separate; it is very difficult to resolve all the hydrocarbons in gasoline, plus, given the overwhelming presence of C8 species, its tough to see a difference:
Next, a little heavier - JP-5 fuel, which is a high flashpoint Navy jet fuel:
Next, JP-5 fuel doped with some n-c8. Since chromatography is a comparative technique at heart, the addition of the notable c8 spike lets you recognize the rest of the hydrocarbons:
Next, road diesel fuel. Since all of these fuels were tested by the same chromatography method, it is valid to compare the times it takes on the scale for the various compounds to elude on these chromatograms versus those of the other fuels:
Next, road diesel fuel doped with some n-c8 for numbering purposes. Fortunately this method doesnt separate the isomers of the various hydrocarbons, so we get real nice distinct peaks where each one is:
And, once again, since it is a comparative technique and the same method was always used... here is the general location where your C8 molecules come through:
Enjoy!
[ July 18, 2006, 04:59 PM: Message edited by: JHZR2 ]





